In code implementation 1 of the previous phase, we used kermer's idea to obtain two data sets through conditional statements. One set is a 20bp fragment containing all gene sequences of a single strain, and the other set is a genome that appears in all (different genomes) of the same strain at the same time. In this phase, we will get candidate primers that can meet the basic requirements.
1, The percentage of kermer per strain was calculated
In the last issue, our ideas were idealized and used the ideas of "all" and "None" to solve the problem. In fact, my preliminary results could not meet my requirements. Therefore, we made corrections in today's article. First, we counted the proportion of kermer sequences of corresponding strains in the total kermer.
The code is as follows:
f = open("Alpha_kermer.txt","a+")
f.write("kermer" + "\t" + "genome" + "\t" + "used_genome" + "\t" + "kermer_number" + "\t" + "frequency" + "\t" + "freq_percent" + "\n")
k_number = 0
for key,value in bb:
f.write(key + "\t" + str(i) + "\t" + str(ii) + "\t" + str(len(bb)) + "\t" + str(value) + "\t" + str('%.2f'%(value * 100 / ii)) + "%" + "\n")
f.close()The results are shown as follows. From left to right, there are kermer sequence, number of genomes, number of genomes used this time, total number of kermer, number of occurrences of this kermer, and proportion of kermer.
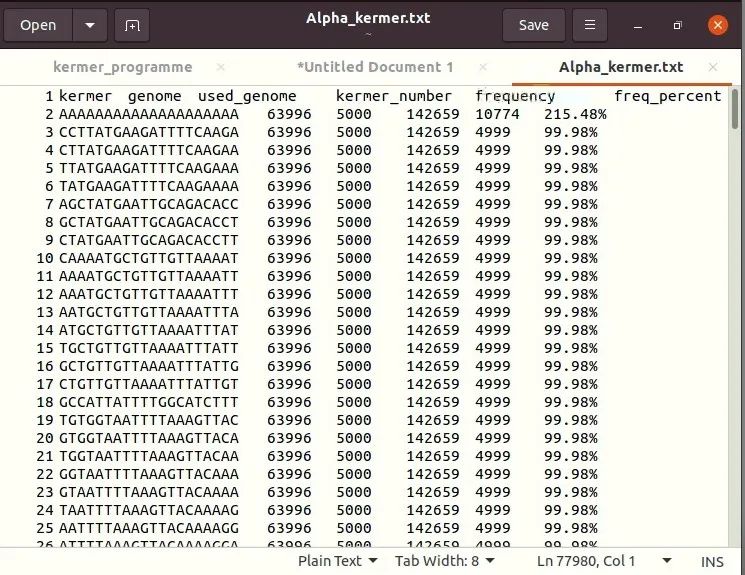
The figure above shows some data of Alpha strain kermer. For the results of other strains, use search to replace the "Alpha" above with the corresponding strain name.
2, The proportion of single strain kermer in other strains kermer was calculated
f2 = open("Alpha_kermer.txt","r")
f4 = open("Beta_kermer.txt","r")
f6 = open("Gamma_kermer.txt","r")
f8 = open("Delta_kermer.txt","r")
Alpha_kermer = []
i = 0
for line in f2.readlines():
i += 1
if i > 1:
Alpha_kermer.append(line.replace("\n",""))
f2.close()
Beta_kermer = []
i = 0
for line in f4.readlines():
i += 1
if i > 1:
Beta_kermer.append(line.replace("\n",""))
f4.close()
Gamma_kermer = []
i = 0
for line in f6.readlines():
i += 1
if i > 1:
Gamma_kermer.append(line.replace("\n",""))
f6.close()
Delta_kermer = []
i = 0
for line in f8.readlines():
i += 1
if i > 1:
Delta_kermer.append(line.replace("\n",""))
f8.close()
f9 = open("Alpha_in_Beta_Gamma_Delta.txt","a+")
f9.write("A_kermer" + "\t" + "per_A_kermer" + "\t" + "per_B_kermer" + "\t" + "per_G_kermer" + "\t" + "per_D_kermer" + "\n")
for i in range(len(Alpha_kermer) - 1):
Alpha = Alpha_kermer[i].split("\t")
m = 0
for ii in range(len(Beta_kermer) - 1):
Beta = Beta_kermer[ii].split("\t")
if Alpha[0] == Beta[0]:
f9.write(Alpha[0] + "\t" + Alpha[5] + "\t" + Beta[5] + "\t")
else:
m += 1
if m == (len(Beta_kermer) - 1):
f9.write(Alpha[0] + "\t" + Alpha[5] + "\t" + "0.00%" + "\t")
m = 0
for ii in range(len(Gamma_kermer) - 1):
Gamma = Gamma_kermer[ii].split("\t")
if Alpha[0] == Gamma[0]:
f9.write(Gamma[5] + "\t")
else:
m += 1
if m == (len(Gamma_kermer) - 1):
f9.write("0.00%" + "\t")
m = 0
for ii in range(len(Delta_kermer) - 1):
Delta = Delta_kermer[ii].split("\t")
if Alpha[0] == Delta[0]:
f9.write(Delta[5] + "\n")
else:
m += 1
if m == (len(Delta_kermer) - 1):
f9.write("0.00%" + "\n")
f9.close()The results are shown in the following figure. From left to right are the kermer sequence of Alpha and the proportion of its kermer in Alpha, Beta, Gamma and Delta.
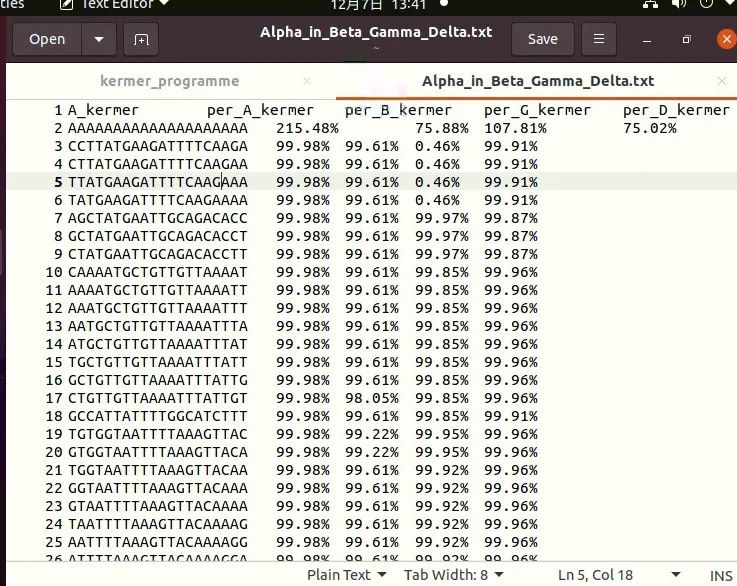
The figure above shows some data of Alpha strain kermer, and the results of other strains are similar.
3, Screening highly specific kermer
The screening method was kermer with high frequency in specific strains and low frequency in other strains.
f9 = open("Alpha_in_Beta_Gamma_Delta.txt","r")
Alpha = []
i = 0
for line in f9.readlines():
i += 1
if i > 1:
Alpha.append(line.replace("\n",""))
f9.close()
f9_1 = open("Alpha_little_in_Beta_Gamma_Delta.txt","a+")
f9_1.write("A_kermer" + "\t" + "per_A_kermer" + "\t" + "per_B_kermer" + "\t" + "per_G_kermer" + "\t" + "per_D_kermer" + "\n")
for i in range(len(Alpha) - 1):
Alpha_little = Alpha[i].split("\t")
if (float(Alpha_little[1].replace("%","")) > 95) & (float(Alpha_little[2].replace("%","")) < 5) & (float(Alpha_little[3].replace("%","")) < 5) & (float(Alpha_little[4].replace("%","")) < 5):
f9_1.write(Alpha[i] + "\n")
f9_1.close()The results are shown in the figure below:
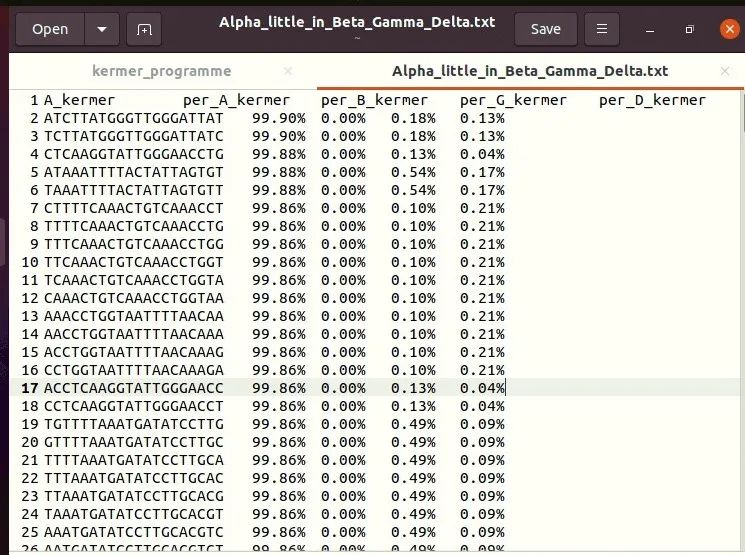
4, Screening candidate primers
Taking the first sequence of each strain as the index template, divide it into 20 BP kermer fragments into the array, then the first element index is 0, the second element index is 1, and the nth element index is n-1, then the size of the product amplified by the primer should be between 75-250 BP, Therefore, the relationship between the index values of Forward Primer and Reverse Primer is: 75 < FM - RN + 20 < 250, where m and N are the index values of pre primer and post primer respectively.
Another consideration for primer screening is the Tm value of the sequence. The annealing temperature during PCR amplification is closely related to the Tm value. The Tm value is called melting temperature (Tm), which refers to the temperature when the absorbance value increases to half of the maximum value, which is called the DNA unwinding temperature or melting point. Generally speaking, Tm value is the melting temperature of DNA and the temperature when the UV absorption reaches half of the maximum value during DNA denaturation. Different sequences of DNA have different Tm values. The higher the C-G content in DNA, the greater the Tm value. When the nucleotide length is less than or equal to 20bp, the calculation formula is Tm = 4 ℃ (G + C)+ 2 ℃ (A + T). In the formula, A, G, C and t represent the number of bases. The general qpcr amplification procedure is as follows:
Pollution prevention stage: 50 degrees and 2min
Pre denaturation stage: 95 ℃ 10min
Denaturation and extension stage: 95 ° 10s, 60 ° 40s, fluorescence signal recorded, 40 cycles
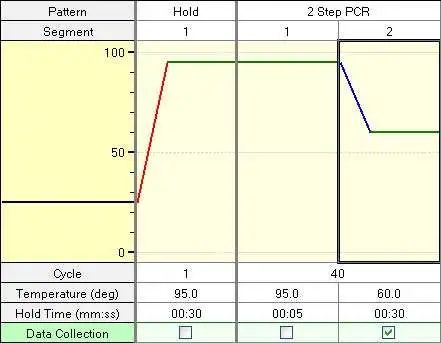
Tm value is usually about 2 degrees higher than the annealing temperature, that is, about 62 degrees. Generally, we set a relatively wide range, such as 50 degrees to 70 degrees.
In addition, the primer requires that the 5 'end start with "A" or "T", and the 3' end ends with "C" or "G", and there can be no continuous "A" or "G" or "C" or "T" in the sequence. The python Programming language to complete the above requirements is as follows:
Alpha_only = []
f9_1 = open("Alpha_little_in_Beta_Gamma_Delta.txt","r")
n = 0
for line in f9_1.readlines():
n += 1
if n > 1:
Alpha_element = line.replace("\n","").split("\t")
Alpha_only.append(Alpha_element[0])
f9_1.close()
AA = len(Alpha_only)
Alpha0 = []
i = 0
a = ""
f = open("/home/lxh/Alpha.fasta","r")
for line in f.readlines():
if ">" in line:
i += 1
Alpha0.append(a)
a = ""
if ">" not in line:
a += line.replace("\n","")
del Alpha0[0]
f.close()
Alpha0_kermer = []
for mm in range(len(Alpha0[0]) - 20):
Alpha0_kermer.append(Alpha0[0][mm:(20 + mm)])
index_Alpha = {}
for i in range(AA - 1):
index = Alpha0_kermer.index(Alpha_only[i])
index_Alpha[Alpha_only[i]] = index
index_sort_A = sorted(index_Alpha.items(),key = lambda x:x[1],reverse = False)
Alpha_num = []
for x,y in index_sort_A:
Alpha_num.append(y)
f_primer = open("Alpha_primer.txt","a+")
NN = len(Alpha_num)
for e in range(NN - 1):
mgb = ''.join(Alpha0_kermer[Alpha_num[e]])
A = mgb.count("A")
G = mgb.count("G")
C = mgb.count("C")
T = mgb.count("T")
Tm1 = 4 * (G + C) + 2 * (A + T)
if (Tm1 < 70) & (Tm1 > 50) & ("AAA" not in mgb) & ("GGG" not in mgb) & ("CCC" not in mgb) & ("TTT" not in mgb) & ((mgb[0] == "A") or (mgb[0] == "T")) & ((mgb[19] == "C") or (mgb[19] == "G")):
number = 0
for r in range((e + 1),NN):
if ((Alpha_num[r] - Alpha_num[e]) > 55) & ((Alpha_num[r] - Alpha_num[e]) < 230):
mgb = ''.join(Alpha0_kermer[Alpha_num[r]])
A = mgb.count("A")
G = mgb.count("G")
C = mgb.count("C")
T = mgb.count("T")
Tm2 = 4 * (A + T) + 2 * (G + C)
if (Tm2 < 70) & (Tm2 > 50) & (abs(Tm2 - Tm1) < 10) & ("AAA" not in mgb) & ("GGG" not in mgb) & ("CCC" not in mgb) & ("TTT" not in mgb) & ((mgb[0] == "C") or (mgb[0] == "G")) & ((mgb[19] == "A") or (mgb[19] == "T")):
number += 1
f_primer.write(Alpha0_kermer[Alpha_num[r]] + "\n")
if number > 0:
f_primer.write("<" + Alpha0_kermer[Alpha_num[e]] + ">" + "\n" + str(number) + "\n")
f_primer.close()
If there are few or no candidate primer sequences, the requirements for the above parts can be relaxed appropriately.
One of the candidate primers for specific detection of Alpha strain is shown below:
Forward_Primer: 5'-ACAGTCATGTACTTAACATC-3'
Reverse_Primer: 5'-TTACCGATATCGATGCACTG-3'
The amplified sequence (168bp) was:
ACAGTCATGTACTTAACATCAACCATATGTAGTTGATGACCCGTGTCCTATTCACTTCTATTCTAAATGGTATATTAGAGTAGGAGCTATAAAATCAGCACCTTTAATTGAATTGTGCGTGGATGAGGCTGGTTCTTAATCACCCATTCAGTGCATCGATATCGGTAA
5, Primer validation - specificity test
-
Species specific test - NCBI blast
national center for biotechnology information, or NCBI for short, is one of the most important databases in life science. It contains published articles, various sequences, structures, etc., as well as its additional functions, such as comparison, annotation information, classification information, SNP analysis, primer online design, etc. we use the blast function of NCBI to compare primers to see if they will be mapped to other databases Species.
The comparison results are all new crown genome, with good specificity! The most basic and important part of a R & D project for detecting new crown mutant kit has been completed.
