main points
-
Megahit introduction
-
Basic assembly principle of Megahit
-
Installation and use of Megahit
-
Megahit actual combat
hello, hello everyone. Today we bring you a super detailed installation and application tutorial about Megahit, a macro genome assembly tool.
We will continue to bring you a series of articles on the analysis of biomedical big data. You are welcome to pay attention and see the articles in a more timely manner.
1, Megahit introduction
Megahit is an ultra fast metagenome de novo assembly tool developed by HKU-BGI. Compared with other genome assembly software, megahit has great advantages in computing time and memory consumption. It is suitable for the assembly of samples in complex environments such as soil and the mixed assembly of a large number of samples [1,2].
2, Basic assembly principle of megahit
Megahit's algorithm is DBG method (de Bruijn graph) based on kmer iteration. Original link: https://doi.org/10.1093/bioinformatics/btv033 , those who are interested can expand their reading.
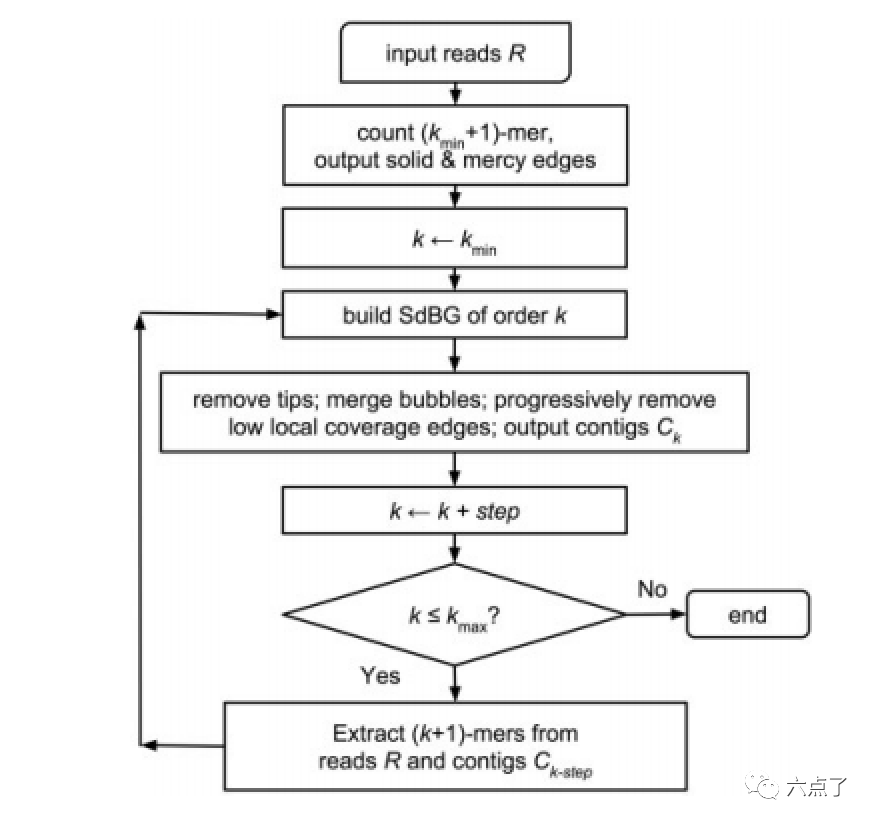
Edit switch to full width
As shown in the figure above, Megahit first divides the read to be assembled into smaller k-mer fragments and constructs a simplified version of SDBG (successful de Bruijn graphs) [3].
There are three dynamic variable parameters: the minimum kmer length Kmin, the maximum kmer length Kmax, and the Step step increased by kmer in each iteration.
1) Megahit first uses the smallest kmer: Kmin to build a simplified de brujin graph, then removes the tips on the de brujin graph, merges the bubbles, and removes the edges with low coverage (similar to the processing of SOAPdenovo mentioned earlier), so as to build the conting: CK under the kmer;
2) Increase the current kmer to kmer+step, then divide the read and the previously assembled conting (CK step) into kmer according to the current kmer size, continue the previous SdBG composition operation, and terminate until the final kmer size reaches Kmax;
3) Conting: CK obtained in the last iteration is the final assembly result.
3, Installation and use of megahit
3.1 installation
Megahit's GitHub link is: https://github.com/voutcn/megahit . The installation of megahit is relatively simple. It can be installed in the following three ways. Users can choose according to their actual situation.
1. Conda installation:
conda install -c bioconda megahit
2. Download binaries directly:
wget https://github.com/voutcn/megahit/releases/download/v1.2.9/MEGAHIT-1.2.9-Linux-x86_64-static.tar.gz tar zvxf MEGAHIT-1.2.9-Linux-x86_64-static.tar.gz cd MEGAHIT-1.2.9-Linux-x86_64-static/bin/ #You can see the executable megahit in the directory
3. Source code compilation and installation:
git clone https://github.com/voutcn/megahit.git cd megahit git submodule update --init mkdir build && cd build cmake .. -DCMAKE_BUILD_TYPE=Release # add - DCMAKE_INSTALL_PREFIX=MY_PREFIX if needed make -j4 make simple_test # will test MEGAHIT with a toy dataset # make install if needed
When B8 +. Zip = and B8 +. Zip = are used to decompress the software, we need to use the following two functions: B8 +. Zip = and B8 +. Zip = to decompress the software.
3.2 instructions for use
Basic usage:
megahit [options] {-1 <pe1> -2 <pe2> | --12 <pe12> | -r <se>} [-o <out_dir>]Detailed explanation of parameters:
#Parameters must be entered -1 <pe1> comma-separated list of fasta/q paired-end #read1 Sequence file for -2 <pe2> comma-separated list of fasta/q paired-end #read2 Sequence file for --12 <pe12> comma-separated list of interleaved fasta/q paired-end files # Staggered double ended PE sequence -r/--read <se> comma-separated list of fasta/q single-end files# Sequence file of single ended SE #Basic parameters: --min-count <int> minimum multiplicity for filtering (k_min+1)-mers [2] #Filtered minimum coincidence bp number --k-list <int,int,..> comma-separated list of kmer size #Set a fixed list of kmer iterations. Comma separated, must be odd #Another kmer Iteration list setting method --k-min <int> minimum kmer size (<= 255), must be odd number [21] #Set minimum kmer size,Should be less than 255, must be odd --k-max <int> maximum kmer size (<= 255), must be odd number [141] #Set maximum kmer size,Should be less than 255, must be odd --k-step <int> increment of kmer size of each iteration (<= 28), must be even number [12] #The interval size should be less than or equal to 28 and must be an even number #Advanced parameters --no-mercy do not add mercy kmers --bubble-level <int> intensity of bubble merging (0-2), 0 to disable [2] #bubble Fusion strength --merge-level <l,s> merge complex bubbles of length <= l*kmer_size and similarity >= s [20,0.95] # Merge length<= l*kmer_size And similarity>= s Complex bubble --prune-level <int> strength of low depth pruning (0-3) [2] # Trim strength for low depth areas --prune-depth <int> remove unitigs with avg kmer depth less than this value [2]# If kmer If the average depth is lower than this value, the is removed unitigs --disconnect-ratio <float> disconnect unitigs if its depth is less than this ratio times #If the depth is less than this ratio multiplied by its depth, it will be disconnected unitigs --low-local-ratio <float> remove unitigs if its depth is less than this ratio times # If the depth is less than this ratio times the adjacent unitigs The average depth is removed unitigs --max-tip-len <int> remove tips less than this value [2*k] #Remove less than this value tips --cleaning-rounds <int> number of rounds for graph cleanning [5] # --no-local disable local assembly #Partial assembly is prohibited #Hardware parameters -m/--memory <float> max memory in byte to be used in SdBG construction # Set the memory size in byte,If less than 1, it is the proportion of the maximum memory of the machine -t/--num-cpu-threads <int> number of CPU threads #Number of threads #Output parameters -o/--out-dir <string> output directory [./megahit_out] #Output directory --out-prefix <string> output prefix (the contig file will be OUT_DIR/OUT_PREFIX.contigs.fa) #Prefix of output result file --min-contig-len <int> minimum length of contigs to output [200] #Set output contig minimum value --keep-tmp-files keep all temporary files #Keep all intermediate files --tmp-dir <string> set temp directory # Set temporary directory
3.3 software operation
#Double ended megahit -1 pe_1.fq -2 pe_2.fq -o out # 1 paired-end library #Cross double ended megahit --12 interleaved.fq -o out # one paired & interleaved paired-end library # Single ended and double ended hybrid megahit -1 a1.fq,b1.fq,c1.fq -2 a2.fq,b2.fq,c2.fq -r se1.fq,se2.fq -o out # 3 paired-end libraries + 2 SE libraries # use megahit_core Get one in the middle kmer of fasta file megahit_core contig2fastg 119 out/intermediate_contigs/k119.contig.fa > k119.fastg # get FASTG from the intermediate contigs of k=119
4, megahit actual combat
Now let's find the sequencing data of Wuhan Xinguan and practice it.
4.1 data download
Download sequencing data:
wget http://www.sixoclock.net/resources/data/NGS/SARS-COV-2/RNA_Seq/nCoVR.WH-100K_1.fastq.gz wget http://www.sixoclock.net/resources/data/NGS/SARS-COV-2/RNA_Seq/nCoVR.WH-100K_2.fastq.gz
4.2 operation command
Here, we redirect the standard output and standard error of the program to the corresponding log and err files respectively.
./megahit -1 nCoVR.WH-100K_1.fastq.gz -2 nCoVR.WH-100K_2.fastq.gz -o out/ 1> log 2>err
4.3 output results
Our test data here is relatively small, so we can run very quickly. The specific results are shown in the figure below. We can see that many intermediate result files have been generated, including final contigs. FA is the assembly result.
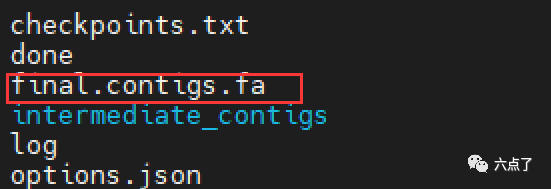
In addition, our sixoclock official website encapsulates megahit software based on CWL (common workflow language), and the sixbox software we developed can run the software quickly.
Those interested in sixbox can go to the official website at six o'clock https://docs.sixoclock.net/clients/sixbox-linux.html Yes.
The specific operation steps are as follows:
1) Download the CWL source code # sixbox pull 321daf5b-e0cf-4d3e-96e1-8773a0a71db0 # or download megahit on the official website of six o'clock cwl[1] .
2) Download data
wget http://www.sixoclock.net/resources/data/NGS/SARS-COV-2/RNA_Seq/nCoVR.WH-100K_1.fastq.gz wget http://www.sixoclock.net/resources/data/NGS/SARS-COV-2/RNA_Seq/nCoVR.WH-100K_2.fastq.gz
3) Use sixbox to generate parameter template file (YAML) and configure yaml file
sixbox run --make-template ./megahit.cwl > megahit.job.yaml vim megahit.job.yaml # Edit parameter profile, replace or set parameters to realize personalized analysis
You can paste the following sample content directly into megahit job. yaml
threads: 2 # default value of type "int". pe2: # type "File" (optional) class: File path: http://www.sixoclock.net/resources/data/NGS/SARS-COV-2/RNA_Seq/nCoVR.WH-100K_2.fastq.gz pe1: # type "File" (optional) class: File path: http://www.sixoclock.net/resources/data/NGS/SARS-COV-2/RNA_Seq/nCoVR.WH-100K_1.fastq.gz outpre: "final" # default value of type "string". outdir: "./megahit_out" # default value of type "string". min_count: 2 # default value of type "int". k_list: "21,29,39,59,79,99,119,141" # default value of type "string".
4) Run with sixbox
sixbox run ./megahit.cwl ./megahit.job.yaml #Or specify the output directory sixbox run --outdir /home/path ./megahit.cwl ./megahit.job.yaml
Run the results to see the current directory or the specified output directory and output the assembly results.
So far, megahit's actual combat experience is basically over.
The above brings you the basic principle knowledge of Megahit, a macrogenome assembly tool, and the detailed operation process.
If you are interested in the content related to biomedical health big data, you can also continue to pay attention to us. To explore the introduction and use of more biomedical big data analysis tools and software, please see It's six o'clock[4].
References
[1] megahit.cwl: https://www.sixoclock.net/application/pipe/321daf5b-e0cf-4d3e-96e1-8773a0a71db0
[2] Li, D., Luo, R., Liu, C.M., Leung, C.M., Ting, H.F., Sadakane, K., Yamashita, H. and Lam, T.W., 2016. MEGAHIT v1.0: A Fast and Scalable Metagenome Assembler driven by Advanced Methodologies and Community Practices. Methods.
[3] Bowe,A. et al. (2012) Succinct de Bruijn Graphs. In: B., Raphael and J., Tang
(eds.) Algorithms in Bioinformatics. Springer, Berlin, pp. 225–235
[4] At six o'clock, the official website: http://www.sixoclock.net
Recommended reading
•This paper explains the principle and practice of genome denovo assembly in detail
•Start! The CWL Chinese course of student information analysis -- Introduction and practice is coming
•Configuration and use of phase III Gatk snp calling process
•Process operation of GATK best practices in phase IV