reference resources https://github.com/cmks/DAS_Tool
DAS: dereplication, aggregation and scoring strategy
DAS Tool can integrate the bins obtained from different macrogenomes, obtain more high-quality, high integrity and non redundant bins, and better show the differences between strain variation microbial strains.
When input ting DAS Tool, you can select as many binning methods as possible. Even some binning methods that only obtain few high-quality bins may also obtain some bins ignored by other methods.
ABAWACA performs a hierarchical clustering on tetranucleotide frequencies and differential coverage, and takes marker genes into account. CONCOCT uses Gaussian mixture models and tetranucleotides frequencies with differential coverage9 . MaxBin 2 is based on an expectation-maximization algorithm and uses tetranucleotides, differential coverage and marker genes13. MetaBAT applies a k-medoid clustering on tetranucleotide frequencies and differential coverage. (quoted from) Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy | Nature Microbiology)
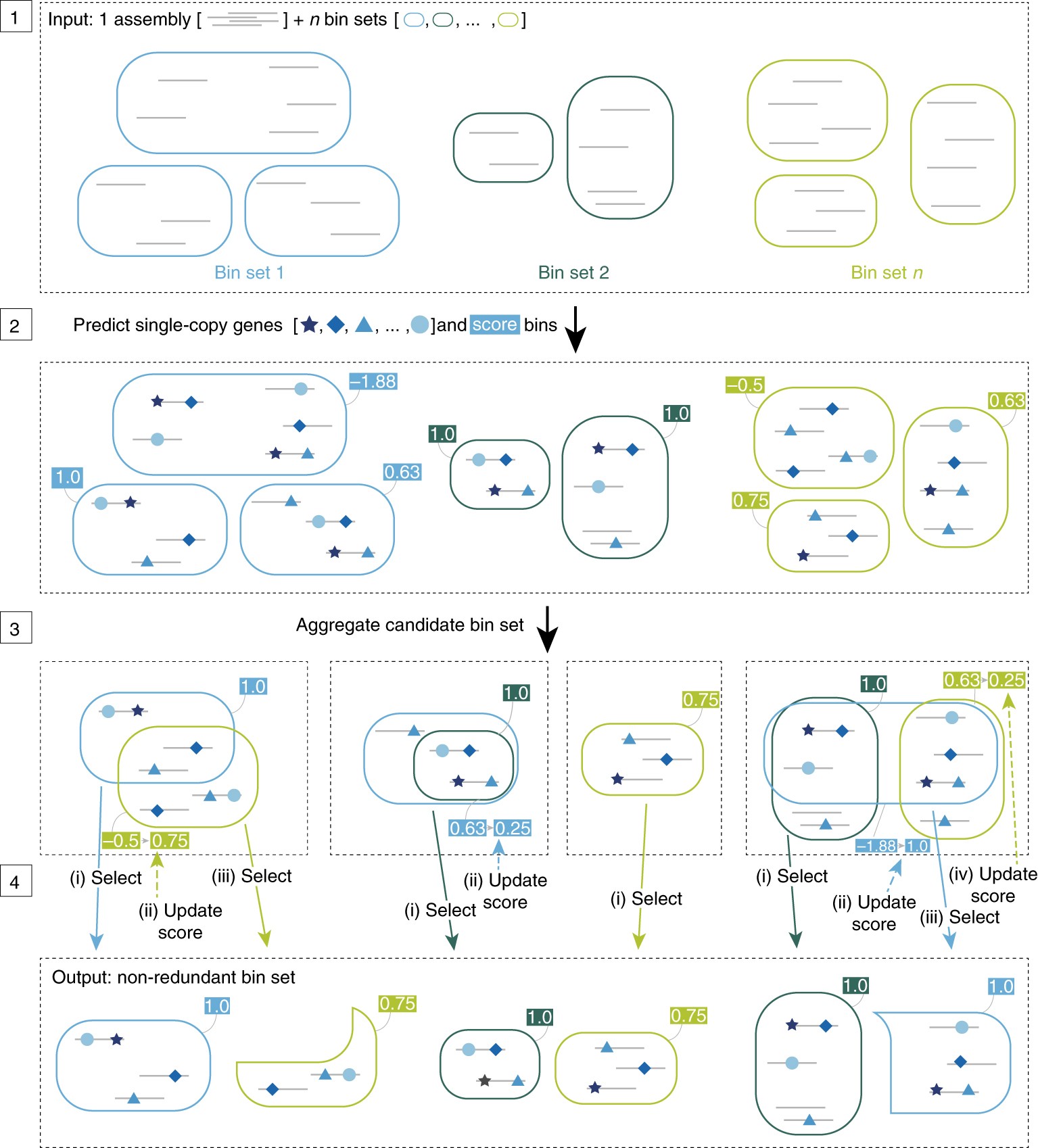
The core idea of DAS is the iteration of judging bin quality based on single copy gene score.
Step 1: the input file of the DAS tool includes the scaffolds sequence (represented by gray lines) in the splicing results and the bins set obtained from different binning tools (rounded rectangles of the same color represent bins obtained by the same binning method);
Step 2: predict the single copy gene of scaffold in each bin (represented by blue shape) and score;
Step 3: in all results, merge the same bins as an alternative set of this bin;
Step 4: iteratively select high score bins and update the scores of candidate bins in the rest of the set. If the scores are the same, select the bin with higher scaffold N50 value. N50 value: the minimum conting length required to cover 50% of the genome
Several reads are obtained by sequencing. These reads are spliced. If they can be spliced completely, the sequence without gap in the middle is called conting, which means continuous. If there is a gap in the middle, but the length of the gap can be known, such a sequence is called scaffold, The meaning of scaffold (discontinuous). Arrange conting and scaffold from long to short, and then add them. When they are just added to 50% of 1M, that is, 500k, the length of that conting or scaffold is called Contig N50 and Scaffold N50. Obviously, the larger this value is, the better the assembly quality is
That is, count down from the longest segment to the segment with a length of half of the total length. The longer the last segment is counted, the more long segments, and the better the quality of the final assembly.
Quoted from What do n50 and N90 mean in genome sequencing_ Mr tomato egg blog - CSDN blog_ What does n50 mean
The final output includes non redundant high score bins (score greater than threshold t) predicted from different input files.
CheckM first constructs the evolutionary tree of the genome based on the complete sequenced bacterial genome as the reference genome, Construct a single copy gene set for each lineage (which can be understood as a species) (single copy genes, SCGs, why single copy? Because it can evaluate the degree of genomic mixing, pollution, etc.). When using, build a tree with Bin and the reference genome, find the reference species of Bin based on the evolutionary relationship, and then calculate two important indicators in combination with the single copy gene set of the reference species. Completeness, Bin gene and pair Whether the number of genes is complete compared with SCGs, the value is [0100%]. The larger the value, the better the quality of Bin; Contamination, pollution degree, Bin gene contains SCGs of multiple species, that is, the degree of multiple species in a Bin. The value is [0100%]. The smaller the value, the better the quality of Bin.
Practical operation
DAS_Tool -i methodA.scaffolds2bin,...,methodN.scaffolds2bin
-l methodA,...,methodN -c contigs.fa -o myOutput
-i, --bins Comma separated list of tab separated scaffolds to bin tables.
-c, --contigs Contigs in fasta format.
-o, --outputbasename Basename of output files.
-l, --labels Comma separated list of binning prediction names. (optional)
--search_engine Engine used for single copy gene identification [blast/diamond/usearch].
(default: usearch)
--write_bin_evals Write evaluation for each input bin set [0/1]. (default: 1)
--create_plots Create binning performance plots [0/1]. (default: 1)
--write_bins Export bins as fasta files [0/1]. (default: 0)
--proteins Predicted proteins in prodigal fasta format (>scaffoldID_geneNo).
Gene prediction step will be skipped if given. (optional)
--score_threshold Score threshold until selection algorithm will keep selecting bins [0..1].
(default: 0.5)
--duplicate_penalty Penalty for duplicate single copy genes per bin (weight b).
Only change if you know what you're doing. [0..3]
(default: 0.6)
--megabin_penalty Penalty for megabins (weight c). Only change if you know what you're doing. [0..3]
(default: 0.5)
--db_directory Directory of single copy gene database. (default: install_dir/db)
--resume Use existing predicted single copy gene files from a previous run [0/1]. (default: 0)
--debug Write debug information to log file.
-t, --threads Number of threads to use. (default: 1)
-v, --version Print version number and exit.
-h, --help Show this message.
-i input the bins results obtained by different binning methods. The file format is tabular scaffolds 2bin file, including tab separated scaffold IDs and bin IDs.
Scaffold_1 bin.01 Scaffold_8 bin.01 Scaffold_42 bin.02 Scaffold_49 bin.03
-l binning method corresponding to - i input file one by one
-c assembled Contag's fasta file
-o output files to the specified folder, including DASTool_summary.txt (output bins and estimation of its quality and integrity); dastool_scaffolds 2bin.txt (output bins and its corresponding scaffolds)
--search_engine
The search method for single copy gene recognition is usearch by default, including blast and diamond
--write_bin_evals
Evaluate each input bin set
--write_bins
Output bins as fasta file
--proteins
fasta format of protein predicted by Prodigal
--score_threshold
Select the threshold for bin