In the era of precision medicine, the most important thing for cancer is to divide and cure. Theoretically, everyone's cancer should be different, but the actual medical reality does not allow us to carry out meticulous scientific research and Exploration on each cancer patient to formulate personalized diagnosis and treatment plans for him. It has been the achievement of the scientific research community in recent decades to distinguish the single organ cancer we talked about before into different sub cancers.
For example, breast cancer, you can see lumA,lumB,basal,HER2 and other subtypes, TNBC can continue to be subdivided into 3~7 subtypes, of course, now that there is a single cell transcriptome data support, cell subtypes will become more and more clear. If we want to integrate multi omics data, the classification will be more complex.
Another example is gastric cancer, which also has four molecular types, as follows:
- ① Epstein Barr virus (EBV) positive tumors account for about 9% of gastric cancer, characterized by high frequency PIK3CA gene mutation and DNA hypermethylation, as well as JAK2, CD274 (also known as PD-L1) and PDCD1LG2 (also known as PD-L2) gene amplification.
- ② Microsatellite instability (MSI): about 22%, showing an increase in repeated DNA sequence mutations, including gene mutations encoding targeted oncogenic signal proteins.
- ③ Gene stable (GS) type: about 20%, its histological variation is diffuse and abundant, and RHOA gene mutation or RHO family GTP ase activated protein gene fusion are common.
- ④ Chromosomal instability (CIN): this kind of tumor accounts for nearly half of gastric cancer, showing significant aneuploidy and local amplification of receptor tyrosine kinase.
Here, share an interesting R package: CancerSubtypes
- https://bioconductor.org/packages/release/bioc/html/CancerSubtypes.html
The article that published this CancerSubtypes package is: (2017). "CancerSubtypes: an R/Bioconductor package for molecular cancer subtype identification, validation, and visualization." https://doi.org/10.1093/bioinformatics/btx378.
It integrates six common molecular typing algorithms for tumor transcriptome sequencing data, including:
- Consensus clustering (CC) (Monti et al., 2003)
- Consensus non-negative matrix factorization (CNMF) (Brunet et al., 2004)
- Integrative clustering (iCluster) (Shen et al., 2009)
- Similarity network fusion (SNF) (Wang et al., 2014)
- Weighted SNF (WSNF) (Xu et al., 2016)
You can see that it has a certain age!
When analyzing TCGA database, you first need to download all the data of 33 cancers of TCGA, especially the expression matrix and clinical phenotype information. Here we recommend downloading it in xena of ucsc: https://xenabrowser.net/datapages/
GDC TCGA Acute Myeloid Leukemia (LAML) (15 datasets) GDC TCGA Adrenocortical Cancer (ACC) (14 datasets) GDC TCGA Bile Duct Cancer (CHOL) (14 datasets) GDC TCGA Bladder Cancer (BLCA) (14 datasets) GDC TCGA Breast Cancer (BRCA) (20 datasets) GDC TCGA Cervical Cancer (CESC) (14 datasets) GDC TCGA Colon Cancer (COAD) (15 datasets) GDC TCGA Endometrioid Cancer (UCEC) (15 datasets) GDC TCGA Esophageal Cancer (ESCA) (14 datasets) GDC TCGA Glioblastoma (GBM) (15 datasets) GDC TCGA Head and Neck Cancer (HNSC) (14 datasets) GDC TCGA Kidney Chromophobe (KICH) (14 datasets) GDC TCGA Kidney Clear Cell Carcinoma (KIRC) (15 datasets) GDC TCGA Kidney Papillary Cell Carcinoma (KIRP) (15 datasets) GDC TCGA Large B-cell Lymphoma (DLBC) (14 datasets) GDC TCGA Liver Cancer (LIHC) (14 datasets) GDC TCGA Lower Grade Glioma (LGG) (14 datasets) GDC TCGA Lung Adenocarcinoma (LUAD) (15 datasets) GDC TCGA Lung Squamous Cell Carcinoma (LUSC) (15 datasets) GDC TCGA Melanoma (SKCM) (14 datasets) GDC TCGA Mesothelioma (MESO) (14 datasets) GDC TCGA Ocular melanomas (UVM) (14 datasets) GDC TCGA Ovarian Cancer (OV) (15 datasets) GDC TCGA Pancreatic Cancer (PAAD) (14 datasets) GDC TCGA Pheochromocytoma & Paraganglioma (PCPG) (14 datasets) GDC TCGA Prostate Cancer (PRAD) (14 datasets) GDC TCGA Rectal Cancer (READ) (15 datasets) GDC TCGA Sarcoma (SARC) (14 datasets) GDC TCGA Stomach Cancer (STAD) (15 datasets) GDC TCGA Testicular Cancer (TGCT) (14 datasets) GDC TCGA Thymoma (THYM) (14 datasets) GDC TCGA Thyroid Cancer (THCA) (14 datasets) GDC TCGA Uterine Carcinosarcoma (UCS) (14 datasets)
The current classification of these cancers is still too rough. It is recommended to use TCGAbiolinks package to obtain the sub group information of each cancer. The code is as follows:
suppressMessages(library(TCGAbiolinks)) suppressMessages(library(tidyverse)) #Download data phe <- as.data.frame(TCGAquery_subtype(tumor = "brca")) table(phe$BRCA_Subtype_PAM50) Basal Her2 LumA LumB NA Normal 192 82 562 209 2 40
You can also simply check the relationship between this subtype and other clinical information:
library(gplots) balloonplot(table( phe$BRCA_Subtype_PAM50, phe$pathologic_stage ))
As follows:
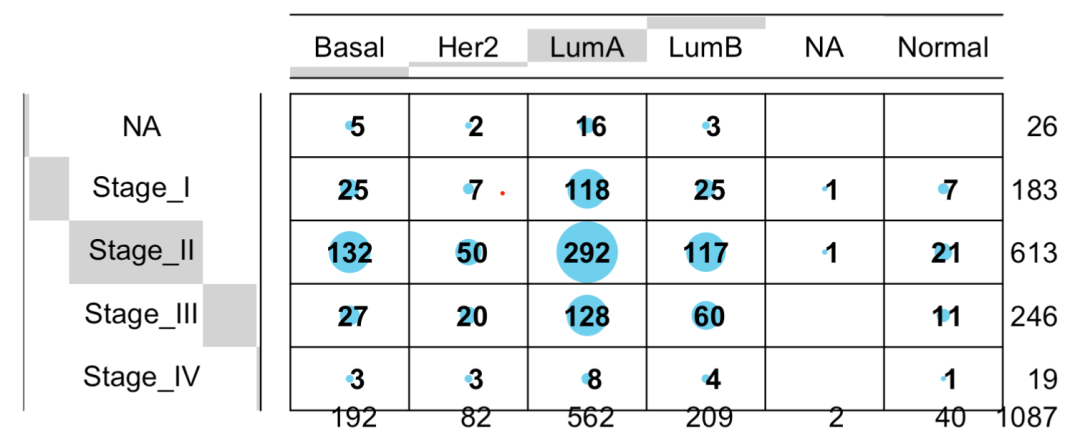
Relationship between this subtype and other clinical information
Because this function obtains too much clinical information, we won't give examples one by one:
> colnames(phe) [1] "patient" "Tumor.Type" [3] "Included_in_previous_marker_papers" "vital_status" [5] "days_to_birth" "days_to_death" [7] "days_to_last_followup" "age_at_initial_pathologic_diagnosis" [9] "pathologic_stage" "Tumor_Grade" [11] "BRCA_Pathology" "BRCA_Subtype_PAM50" [13] "MSI_status" "HPV_Status" [15] "tobacco_smoking_history" "CNV Clusters" [17] "Mutation Clusters" "DNA.Methylation Clusters" [19] "mRNA Clusters" "miRNA Clusters" [21] "lncRNA Clusters" "Protein Clusters" [23] "PARADIGM Clusters" "Pan-Gyn Clusters" >
You can see that R language is really super convenient and suitable for data mining! For example, in my 4-hour TCGA tumor database knowledge map video tutorial, four algorithms are used to build the model:
- Cox (single factor and multi factor) cox model construction and risk forest map of TCGA
- lasso regression Construction of survival model with lasso regression + ROC curve drawing
- Random forest It sounds domineering and is not difficult to use
- Support vector machine Support vector machine, which sounds domineering and is not difficult to use
No matter which algorithm is used, the core is just a few words of code.
Similar to other cancers, you only need to change the acronyms in it. Interested partners can try all cancers once.