2021SC@SDUSC
Shell
Shell is a program, which can be called shell program, which is used for users to interact with the operating system. It is used to distinguish between and core, which is equivalent to a command parser. There are many shells, and several of them are listed here
Bourne SHell(sh)
Bourne Again SHell(bash)
C SHell(csh)
KornSHell(ksh)
zsh
The functions of each shell are not much different. There are some differences in some syntax. The Linux default is bash. Bash is mainly introduced here
bash
Bash command is a superset of SH commands. Most sh scripts can be run under bash. Bash mainly has the following functions
bash function
Record history command: bash can record the previous command and keep it at ~ / bash_ In the history file, only the commands since the last logout and login are saved
Tab key automatic completion: use tab to see the automatic incomplete command or directory i
Alias command alias: alias ll='ls -al 'can be used to set the alias of the command
Work control: some tasks can be run in the background, which is not introduced here
Program script: you can execute shell script files
Wildcards: wildcards can be used when finding related files or executing related commands*
Built in command type: you can use the type command to check whether a command is built in bash
Experiment 11: the use of Bash - Part I
1. Experimental purpose
1) Master the use of Bash command processor
2. Experimental contents
1. Write and execute the first Bash script
2.Bash variable
3.Bash string
4.Bash array
5.Bash notes
6.Bash transfer parameters
3. Experimental environment
Raspberry pie 4B, Ubuntu 21.10. The programming environment of the experiment is Terminal terminal program
4. Experimental steps
1. Write and execute the first Bash script
Open the terminal, create a file through vim command, and create a new file test SH, the extension is sh (SH stands for shell), and the extension will not affect the execution of the script. If you use python to write shell scripts, you'd better use py as the extension.
Enter the following code in the vim editor:
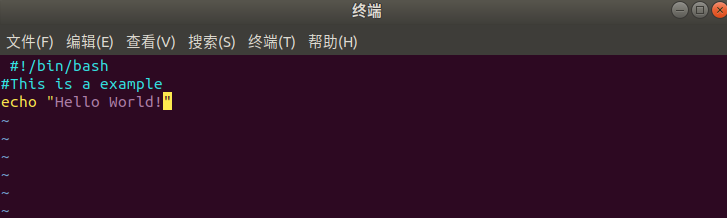 The next step is to run the Bash script
The next step is to run the Bash script
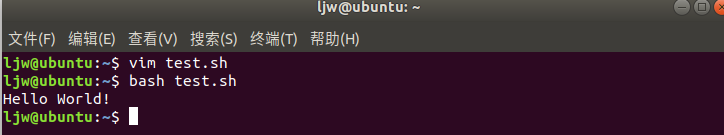
We can directly run the interpreter, and its parameter is the file name of the shell script, such as:
$ /bin/sh test.sh or $bash test sh
The results are as follows:
2.Bash variable
There are two kinds of variables in Bash: environment variables and user-defined variables, which can be understood as global variables and local variables. Before understanding their differences, we need to know the parent program and subroutine. For example, in the current bash, we call it the parent program, and any program executed under this Bash is called the subroutine. The difference between global variables and local variables is that global variables are still valid in subroutines and local variables are only valid in the current program. (note that once the bash of the parent program is exited, both global variables and local variables are invalid. When Bash is opened again, the variable does not exist)
3.Bash string
String is the most commonly used and useful data type in Bash programming. String can use single quotation marks, double quotation marks or no quotation marks.
Single quote declaration string
str='string'
Some limitations of single quote declaration strings:
• any character in the single quotation mark will be output as is, and the variable in the single quotation mark string is invalid;
• a single single quotation mark cannot appear in a single quotation mark string (nor after using an escape character for single quotation marks), but it can appear in pairs and be used as string splicing
Double quote declaration string
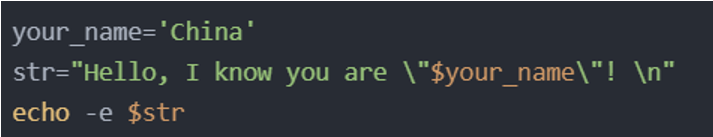
The output result is:
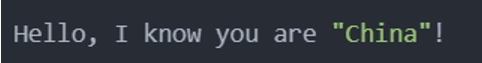
Advantages of double quotation marks:
• there can be variables in double quotation marks
• escape characters can appear in double quotation marks
4.Bash array
There are 2 arrays:
- Index array
- Associative array (Hashmap in java), associating value with key
Declaration array:
#Declare index array declare -a ary #Declare associative arrays declare -a ary
Reference array: ${ary[idx]}
Parentheses must be enlarged
To directly access the array name is to access the first element
[root@localhost ~]# ani[0]=dog
[root@localhost ~]# ani[1]=cat
[root@localhost ~]# echo ani
ani
[root@localhost ~]# echo ani[0]
ani[0]
[root@localhost ~]# echo $ani[0]
dog[0]
[root@localhost ~]# echo ${ani[0]}
dog
[root@localhost ~]# echo ${ani[1]}
cat
Assign a value to an array
-
Single assignment: ary[idx]=val
-
Multiple assignments: ary = (val1, val2...)
[root@localhost ~]# ary=(1 2 3 4 ) [root@localhost ~]# echo $ary 1 [root@localhost ~]# echo ${ary[0]} 1 [root@localhost ~]# echo ${ary[2]} 3 [root@localhost ~]# echo ${ary[1]} 2 [root@localhost ~]# echo ${ary[3]} 4 [root@localhost ~]# echo ${ary[4]} [root@localhost ~]# -
Jump assignment: ary=([0]="aa" [3]="dd")
[root@localhost ~]# ary=([0]="aa" [3]="dd") [root@localhost ~]# echo ${ary[0]} aa [root@localhost ~]# echo ${ary[1]} [root@localhost ~]# echo ${ary[3]} dd
5.Bash notes
Lines beginning with # are comments and are ignored by the interpreter.
Set multi line comments by adding a # sign to each line, like this:
What if you encounter a large piece of code that needs to be annotated temporarily during the development process and cancel the annotation later?
Adding a # symbol to each line is too laborious. You can enclose this section of code to be annotated with a pair of curly braces and define it as a function. If there is no place to call this function, this code will not be executed and achieve the same effect as the annotation.
6.Bash transfer parameters
For example, if I want to output the location information of a gene on the chromosome, I can write the following bash script and save it as gene sh.
The location information of the gene is saved in the script.
#!/bin/bash
START=5000000
END=6000000
SCFID=785
GENE=Cyp6a9
echo The ${GENE} is on Chr${SCFID}:${START}-${END} Then execute it on the command line.
chmod 777 gene.sh ./gene.sh ## The Cyp6a9 is on Chr785:5000000-6000000
However, if the script is not executed only once, but many times. Then each time you target different genes, you have to enter gene SH file, then modify the four parameters, and then save the file. It is very inconvenient and not suitable for batch operation.
At this time, you need to add parameters to the script. In this way, you can enter parameters on the command line every time you execute the script, and there is no need to enter the script file for modification.
Position parameters
The parameters passed in to the corresponding script are represented by the position on the command line. This is the simplest one. As follows, we pass in the four parameters corresponding to START,END,SCFID and GENE.
#!/bin/bash
START=${1}
END=${2}
SCFID=${3}
GENE=${4}
echo The ${GENE} is on Chr${SCFID}:${START}-${END} At this time, when executing, you only need to enter the corresponding parameters on the command line, and the parameters are separated by spaces.
./gene.sh 5000000 6000000 785 Cyp6a9 ## The Cyp6a9 is on Chr785:5000000-6000000
Although this method is simple, the input parameters must be strictly corresponding, and the parameters cannot be left blank or the position can not be reversed, which is very unfriendly to new users of the script.
Parameter name match
Another way to transfer parameters is to use keywords and transfer parameters through keyword matching. In this case, the parameter transfer method is more flexible and does not depend on the parameter position.
#!/bin/bash
usage() {
echo "Usage: ${0} [-s|--start] [-e|--end] [-i|--scfid] [-g|--gene]" 1>&2
exit 1
}
while [[ $# -gt 0 ]];do
key=${1}
case ${key} in
-s|--start)
START=${2}
shift 2
;;
-e|--end)
END=${2}
shift 2
;;
-i|--scfid)
SCFID=${2}
shift 2
;;
-g|--gene)
GENE=${2}
shift 2
;;
*)
usage
shift
;;
esac
done
echo The ${GENE} is on Chr${SCFID}:${START}-${END}At this time, on the command line, you can enter parameters in sequence or in the form of double horizontal lines and full names:
./gene.sh -s 5000000 -e 6000000 -i 785 -g Cyp6a2 ./gene.sh -e 6000000 -i 785 -g Cyp6a2 -s 5000000 ./gene.sh -e 6000000 -i 785 --gene Cyp6a2 -s 5000000 # .....
Of course, if no match is found in the command parameters, an error will be prompted:
./gene.sh dsafda ## Usage: ./gene.sh [-s|--start] [-e|--end] [-i|--scfid] [-g|--gene]
There are two key ways to pass in this parameter. One is to use case statement, and the other is to use shift. Shift is mainly used to move position parameters. Each time it is executed, the first position parameter moves to the right once. Of course, if it is shift n, it means moving n positions to the right every time.